





MetaPlex is a library preparation workflow and read processing toolkit for efficient and accurate COI metabarcoding on Ion Torrent sequencers. At its core, MetaPlex utilizes multiple pairs of uniquely indexed fusion primers which contain Ion sequencing adapters, Ion Xpress Barcodes, and the COI ANML primers, allowing for single-step PCR library preparation of dual-indexed reads.

For a full list of MetaPlex primers and library preparation information, see here
In order to easily process MetaPlex reads in popular analysis platform QIIME2, MetaPlex provides a toolkit capable of the following tasks:
-
Remultiplexing : reorganize dual-indexes to follow ['MultiplexedSingleEndBarcodeInSequence'] format
-
Index jump calculating : calculate the rate at which index jumps occur per sequencing run, and estimate the number of false reads within the total pool as well as each individual sample within the pool
-
Per-sample frequency based filtering : remove false reads from each sample based off the calculated expectancies either provided by the Index Jump calculator, or at user specified levels.
-
All-sample length based filtering : remove sequences below a length threshold from QIIME2 FeatureTable[Frequency] and FeatureData[Sequence] artifacts.
Installation
All MetaPlex tools function properly when installed in a QIIME2 conda environment (> =2021.11)
Install with conda:
conda install -c bioconda metaplex
Or with pip:
pip install metaplex
Remultiplexing
Function: takes dual-indexed reads, trims the 5' and 3' ends of the reads past the indexes, and moves the 3' index to immediately follow the 5' index (i.e. ['MultiplexedSingleEndBarcodeInSequence'] format)


This process allows for proper organized demultiplexing of dual-indexed outputs from single end sequencers (Ion Torrent, PacBio, Nanopore) in QIIME2.
Usage
Example Command Line Call
Metaplex-remultiplex raw_seqs.fastq.gz indexes.csv
Input
The remultiplexing process can start from either a raw unmapped bam file such as what is given by an Ion Torrent sequencer, or a fastq/fastq.gz file. Aside from the sequences, all that is needed is a .csv containing all the index tag sequences used in the sequencing pool. This index map must follow the formatting outlined in the provided example (see below for details).
sequenceFile : path to raw sequence file of type .fastq, .fastq.gz, or .bam
indexFile : path to .csv containing all the index tag sequences that are present in the sequencing pool. This .csv
should be formatted as specified below (See indexes.csv for reference)
Column 1 - 'ID' - two digit identifiers (zero padded if single digit) for the index tags used for sequencing
Column 2 - 'seq' - sequence of the index tag
*** Note that tags placed on the reverse end should be input as revers complements ***
Column 3 - 'orientation' - F if the index was used on the Forward end, or R if on the Reverse end
Output
Remultiplexing produces a single gzipped fastq containing all sequences where both a forward and reverse barcode were found.
This format allows for immediate importing as a QIIME2 artifact of type ['MultiplexedSingleEndBarcodeInSequence']
Return : None
Output : remultiplexed_seqs.fastq.gz
Example Python Import
from metaplex import remultiplexing
remultiplexing.remultiplex('raw_seqs.fastq.gz', 'indexes.csv')
Index Jumping
Function: calculate the rate at which index jumps occur for a single sequencing run, and estimate the number of false reads within the total sample pool, and for each individual sample.
Though this calculation process should be reproducible across many use cases, there are a few key requirements which must be met for it to be accurate.
-
Dual-indexed reads (i.e. an index or barcode placed on both the forward and reverse end) which has been demultiplexed using QIIME2

-
Use of two or more dual-index pairs

-
'Calibrator tags', or the use of at least one forward and one reverse index pair exclusively with each other. While one pair of calibraotr tags is required, we provide support for specifying multiple pairs.



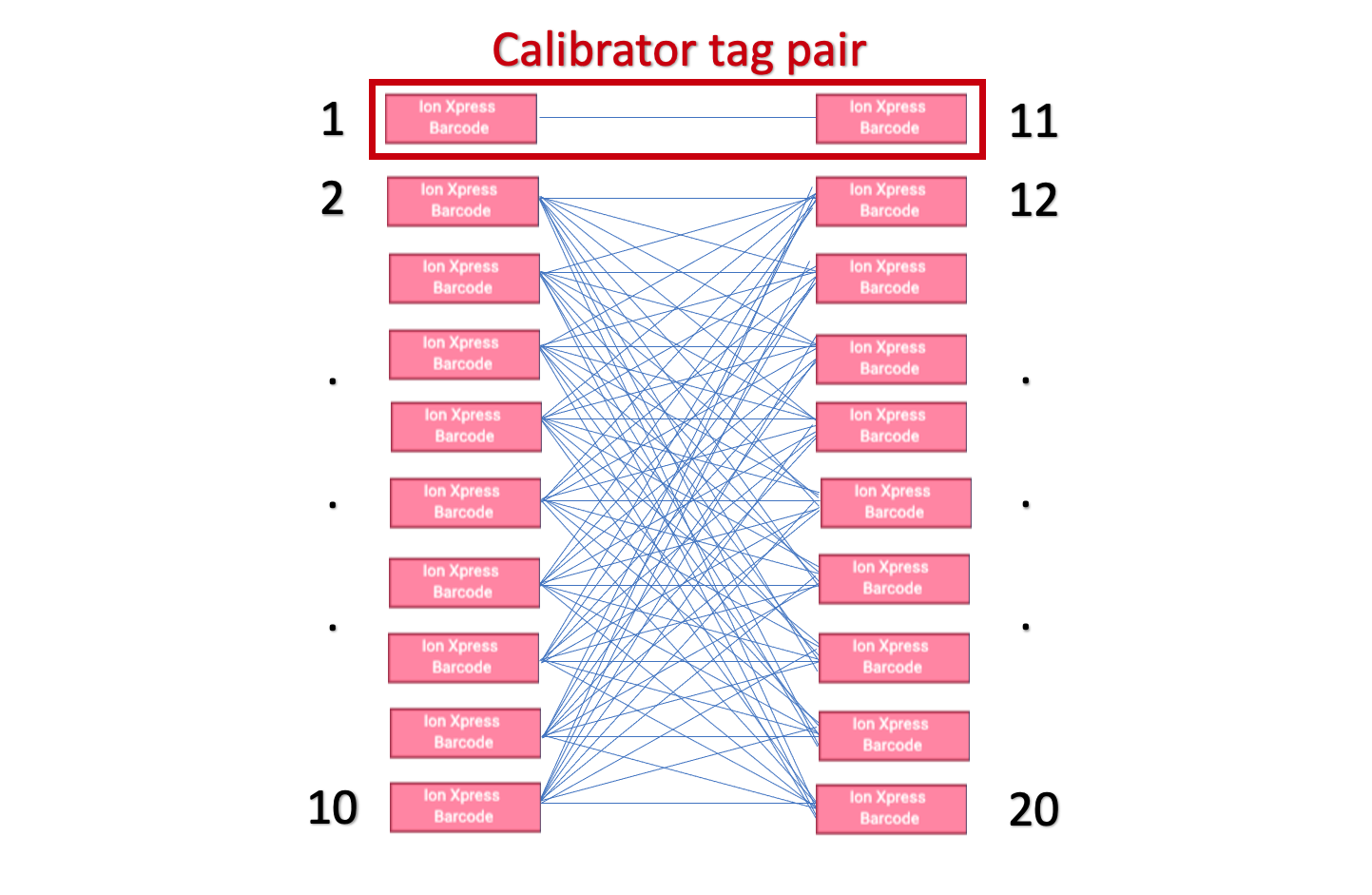
Usage
Example Command Line Call
Metaplex-calculate-IJR demultiplexed_seqs.qza Sample_Map.txt 01,11
Inputs
This workflow requires starting from data which has been demultiplexed utilizing QIIME2. Specifically, we start with a data type SampleData[SequencesWithQuality], though it isn't necessary for the data to have quality values included.
demultiplexed_seqs : path to demultiplexed QIIME2 qza file of data type SampleData[SequencesWithQuality]
sample_map : path to tab delmited QIIME2 sample map file containing ALL possible index combinations
and a True_False indicator column (See example provided in repo)
calibrator_tag_pairs: pairs of calibrator tags, input as either a tuple, or list of tuples, with each
each index being a 2 digit zero padded string.
ex: [('01', '11')] or [('01', '11'), ('02','12')]
Outputs
Return : Integer value of maximum number of index jumps (false reads) expected in a single sample
Output : Expected_False_Reads_Per_Index.csv file containing number of false reads expected in EACH sample
log.txt file containing summary statistics
Example Python Import
from metaplex import index_jump
index_jump.calculate('demultiplexed_seqs.qza', 'Sample_Map.txt', [('01', '11')])
Detailed Index Jump Calculation Workflow
The jump rate of each calibrator tag is calculated by taking the false reads with that tag, and dividing them by the total number of that tag present in the pool.
In this instance a false read is any read which has an F01 tag and not an R11 tag, or any read with an R11 tag and not an F01. The jump rate for the F01 tag and R11 tag are then averaged as a best estimate for the overall rate at which any index jumps occur.
This method of calculating jump rate based off of calibrator tags offers increased accuracy when compared to calculating jump rate based off of just the total false reads within a pool.
In using calibrator tags we are able to detect every instance in which a tag jump occurred in regard to a single index tag. Ultimately this estimate is conservative, as it doesn't take into account indexes which jump but don't actually change the sample assignment, such as the 11 index jumping from an F01R11 sample to another F01R11 sample, nor does it account for the potential for indexes to jump at differential rates.
With this rate, we can then get an estimate for the total number of false reads in the data set by multiplying the total read count by the jump rate. This can be used to give a good estimate of the overall percentage of true and false reads in the data set.
Additionally, we generate a useful table (Expected_False_Reads_Per_Index.csv) with false read estimates calculated per sample taking into account individual tag abundances in the sample pool. For a more in depth look at these calculations take a look at the Jupyter-Notebook provided here.
This csv can then be used to assist in setting a per-sample filtering levels for additional quality control (see PerSampleFiltering).
Per Sample Filtering
Function: Filters reads out of a QIIME2 feature table according to a minimum read count requirement per sample
Usage
Example Command Line Call
Metaplex-per-sample-filter feature_table.qza Expected_False_Reads_Per_Index.csv
Inputs
feature_table : path to QIIME2 feature table
filtering_integer: Either an integer for even filtering across samples, or path to the
Expected_False_Reads_Per_Index.csv output by index_jump.py
Output
Return: Frequency filtered QIIME2 feature table of type FeatureTable[Frequency]
Output: 'freq_filt_table.qza' QIIME2 artifact of type FeatureTable[Frequency]
Example Python Import
from metaplex import per_sample_filtering
per_sample_filtering.per_sample_filter('feature_table.qza', 'Expected_False_Reads_Per_Index.csv')
Here we start from a QIIME2 feature table consisting of 85 uniquely indexed samples. Within each sample are a number of different features with individually recorded frequencies. Below is a histogram of a single sample from our pool with index F02R16, which shows the frequency of each feature within the sample.

After per sample filtering with our input .csv, any feature with a frequency of less than 5 is filtered out of the F02R16 sample. This is carried out for each sample in the pooled feature table according to specifications in the sample map, or at a single depth across all samples as specified by the user.

Length Filtering
Function: Filters reads out of a QIIME2 feature table and rep-seqs file according to a minimum length requirement.
Usage
Example Command Line Call
Metaplex-length-filter feature_table.qza rep_seqs.qza 120
Inputs
feature_table : path to QIIME2 feature table
rep_seqs : path to QIIME2 representative sequences (rep-seqs) file
filtering_integer: An integer threshold for sequence length. All sequences shorter than specified
length will be removed
Output
Return: Frequency filtered QIIME2 feature table of type FeatureTable[Frequency]
Frequency filtered QIIME2 feature table of type FeatureData[Sequence]
Output: 'length_filt_table{length}.qza' QIIME2 artifact of type FeatureTable[Frequency]
'length_filt_seqs{length}.qza' QIIME2 artifact of type FeatureData[Sequence]
Example Python Import
from metaplex import length_filtering
length_filtering.length_filter('feature_table.qza', 'rep_seqs.qza', 120)
In this example, our target amplicon has a length of ~180bp. As seen below, there are a handful of reads which are much shorter than our target.

Here we set our filtering integer to 120 base pairs. After length filtering, any representative sequence with a bp length of less than our specified integer of 120 is filtered out of the sample pool.

This returns both a new feature table, and representative sequences file.
Sample Data
If you are interested in playing with sample data prior to using any of these tools, we provide some test files in this repository.
This sample data is a sub sample of reads from a MetaPlex metabarcoding run on an Ion S5 Prime sequencer.
These are COI reads consisting of 75 True tag combinations, including F01 R11 calibrator tags.
Along with Jupyter Notebooks walking through each MetaPlex tool, we also provide transition notebooks which outline getting your data from one usable step to the next (i.e. remultiplexed seqs to demultiplexed seqs, and demultiplexed seqs to a feature table)
