epiMuller README
About
Author
Jennifer L. Havens
Purpose
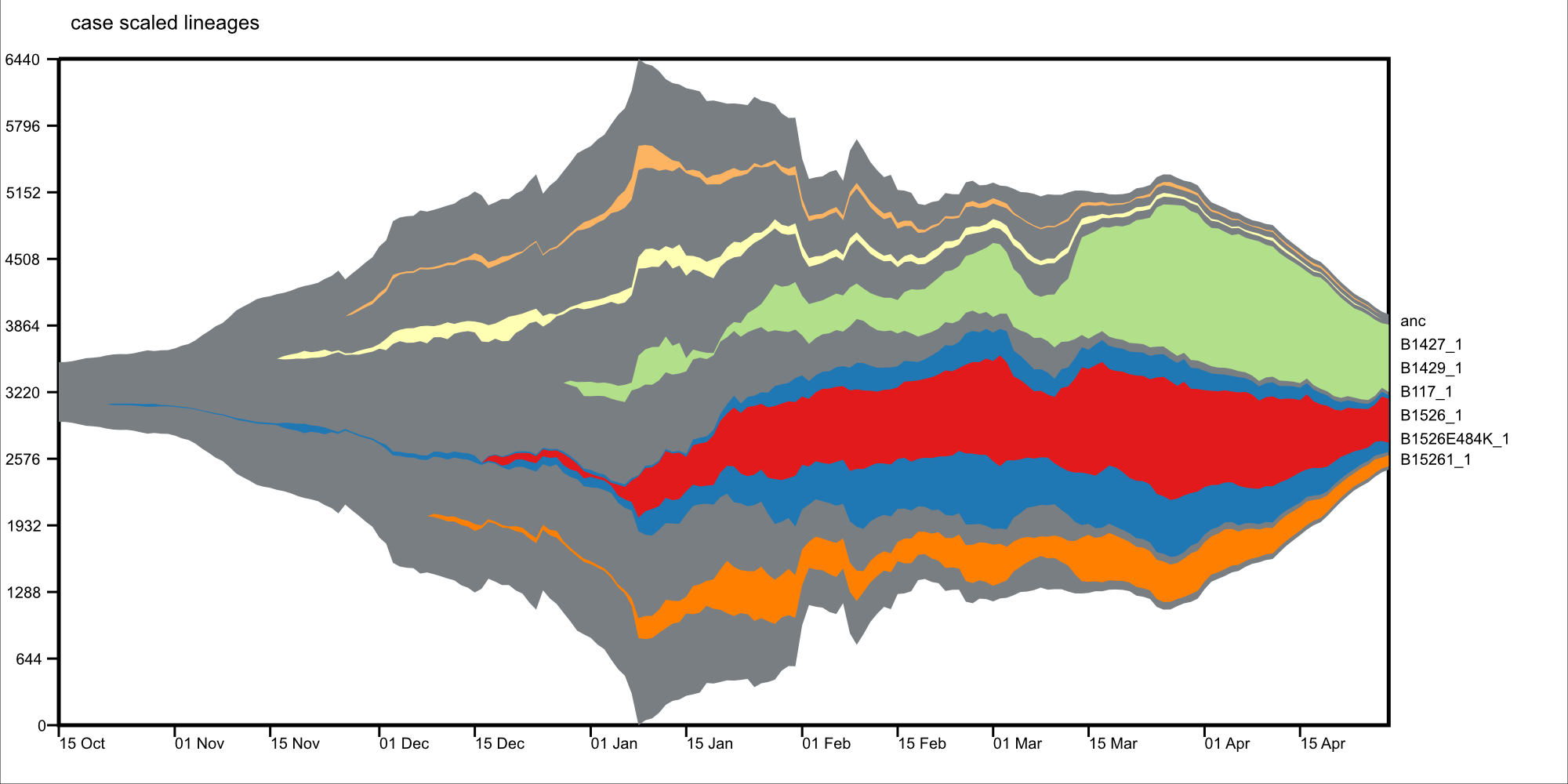
Visualize lineages overtime, with phylogenetic context, based on viral genomes.
Language
Python3
Inputs
timetree, ancestral state reconstruction (Nextstain JSON file or annotated TreeTime nexus file), sample collection dates and, PANGO lineages (optional)
Workflow overview
- epimuller-parse (optional): parse fasta names with 'bar isodate' suffix into usable fasta and metadata files.
-
epimuller: wrapper for epimuller-define and epimuller-draw.
- epimuller-define: assigns samples to clades based on ancestral reconstruction of specified aa mutations or trait (hierarchy), and counts number of samples in a clade withen each time frame (abundance).
- epimuller-draw: plots the frequency clades overtime, as specifed by abundance and hierarchy inputs from epimuller-define.
Source code avaliblity
Documentation avaliblity
Quick start
pip3 install epimuller
epimuller [-h] [-oDir OUTDIRECTORY] -oP OUTPREFIX
(-n INNEXTSTRAIN | -a ANNOTATEDTREE) -m
INMETA [-p INPANGOLIN] [--noPangolin]
[-f TRAITOFINTERSTFILE] [-g GENEBOUNDRY]
[-k TRAITOFINTERSTKEY]
[-mut VOCLIST [VOCLIST ...]] [-t TIMEWINDOW]
[-s STARTDATE] [-e ENDDATE] [-mt MINTIME]
[-min MINTOTALCOUNT] [-c CASES_NAME]
[-l {date,time}] [-lp {Right,Max,Start,End}]
[--WIDTH WIDTH] [--HEIGHT HEIGHT]
[--LEGENDWIDTH LEGENDWIDTH] [--MARGIN MARGIN]
[--FONTSIZE FONTSIZE]
[--LABELSHIFT LABELSHIFT]
SOME EXAMPLES
Examples for full run
To see steps used to prep files for these examples look at scripts/Example_CommandsFromScratch.txt on gitHub.
Visulize default aa mutation list
epimuller \
-n inputData/GISAID_NYCPHL_04_29/02_nextstrainResults \
-m inputData/GISAID_NYCPHL_04_29/gisaid_2021_04_30_00_rename.tsv \
-oDir 03_results_NYCPHL_April29 \
-oP 01_defaultAAList \
-c inputData/CITY_US-NY_NYC_outbreakinfo_epidemiology_data_2021-04-30.tsv
Visulize a trait: lineage
epimuller \
-n inputData/GISAID_NYCPHL_04_29/02_nextstrainResults \
-m inputData/GISAID_NYCPHL_04_29/gisaid_2021_04_30_00_rename.tsv \
-oDir 03_results_NYCPHL_April29 \
-oP 02_pangolin \
-c inputData/CITY_US-NY_NYC_outbreakinfo_epidemiology_data_2021-04-30.tsv \
--traitOfInterstFile traits.json \
--traitOfInterstKey lineage \
-lp Max \
-min 100 \
Visulize your own aa mutation list
epimuller \
-n inputData/GISAID_NYCPHL_04_29/02_nextstrainResults \
-m inputData/GISAID_NYCPHL_04_29/gisaid_2021_04_30_00_rename.tsv \
-oDir 03_results_NYCPHL_April29 \
-oP 03_selectedAA \
-c inputData/CITY_US-NY_NYC_outbreakinfo_epidemiology_data_2021-04-30.tsv \
-mut 'SE484K' 'S*452*' \
-min 50 \
-mt 20
Visulize default aa mutation list with TreeTime input
epimuller \
-a inputData/GISAID_NYCPHL_04_29/06_treetimeDates_aa/timetree.nexus \
-oDir 04_results_NYCPHL_April29 \
-oP defaultAA_treetime \
-m inputData/GISAID_NYCPHL_04_29/gisaid_2021_04_30_00_rename.tsv \
-g data/geneAAboundries.json \
--FONTSIZE 18
Visulize a trait: lineage with TreeTime input
epimuller \
-a inputData/GISAID_NYCPHL_04_29/06_treetimeDates_aa/timetree.nexus \
-oDir 03_results_NYCPHL_April29 \
-oP 05_pangolin_treetime \
-m inputData/GISAID_NYCPHL_04_29/gisaid_2021_04_30_00_rename.tsv \
--traitOfInterstKey lineage \
--noPangolin #do not label with mode of pangolin lineages in clade, label clade with defining lineage only
Known edge cases / featrues to add
Known edge cases which are not correctly dealt with or features I intend to address (eventually). If you run into anything else please let me know with an issue on gitHub.
- feel free to ignore the undefined.svg that gets made - it is related to checking the size of the text to space out labels
- allow combination of aa mutants, not just 1
- define polytomy behavior
- option for user defined col names in metadata
Addtional features
Color
If you would like to specify color for clade: in --parentHierarchy_name file (of epimuller-draw/drawMuller.py input) add col with name: "color" and hex color value (starting with #) for clades you want to specify.
Parse GISAID fasta for metadata
epimuller-parse If you have downloaded sequences from GISAID under the search tab, you can parse out the names into a metadata file (format tested as of 2021-04-30).
epimuller arguments
epimuller [-h] [-oDir OUTDIRECTORY] -oP OUTPREFIX
(-n INNEXTSTRAIN | -a ANNOTATEDTREE) -m
INMETA [-p INPANGOLIN] [--noPangolin]
[-f TRAITOFINTERSTFILE] [-g GENEBOUNDRY]
[-k TRAITOFINTERSTKEY]
[-mut VOCLIST [VOCLIST ...]] [-t TIMEWINDOW]
[-s STARTDATE] [-e ENDDATE] [-mt MINTIME]
[-min MINTOTALCOUNT] [-c CASES_NAME]
[-l {date,time}] [-lp {Right,Max,Start,End}]
[--WIDTH WIDTH] [--HEIGHT HEIGHT]
[--LEGENDWIDTH LEGENDWIDTH] [--MARGIN MARGIN]
[--FONTSIZE FONTSIZE]
[--LABELSHIFT LABELSHIFT]
optional arguments:
-h, --help show this help message and exit
-n INNEXTSTRAIN, --inNextstrain INNEXTSTRAIN
nextstrain results with tree.nwk and
[traitOfInterst].json (default: None)
-a ANNOTATEDTREE, --annotatedTree ANNOTATEDTREE
nexus file name and [traitOfInterst].json (default:
None)
Options for full repot:
-oDir OUTDIRECTORY, --outDirectory OUTDIRECTORY
folder for output (default: ./)
-oP OUTPREFIX, --outPrefix OUTPREFIX
prefix of out files withen outDirectory (default:
None)
Options passed to epimuller-define:
-m INMETA, --inMeta INMETA
metadata tsv with 'strain' and 'date'cols, optional:
cols of trait of interst; and pangolin col named:
'lineage' or 'pangolin_lin' (default: None)
-p INPANGOLIN, --inPangolin INPANGOLIN
pangolin output lineage_report.csv file, if argument
not supplied looks in inMeta for col with
'pangolin_lin' or 'lineage' (default: metadata)
--noPangolin do not add lineage to cade names (default: False)
-f TRAITOFINTERSTFILE, --traitOfInterstFile TRAITOFINTERSTFILE
name of nextstrain [traitOfInterst].json in
'inNextstrain' folder (default: aa_muts.json)
-g GENEBOUNDRY, --geneBoundry GENEBOUNDRY
json formated file specifing start end postions of
genes in alnment for annotatedTree with aa_muts option
(default: None)
-k TRAITOFINTERSTKEY, --traitOfInterstKey TRAITOFINTERSTKEY
key for trait of interst in json file or annotated
tree file for aa with 'mutations' annotation, use
'aa_muts' (default: aa_muts)
-mut VOCLIST [VOCLIST ...], --VOClist VOCLIST [VOCLIST ...]
list of aa of interest in form
[GENE][*ORAncAA][site][*ORtoAA] ex. S*501*, gaps
represed by X (default: None)
-t TIMEWINDOW, --timeWindow TIMEWINDOW
number of days for sampling window (default: 7)
-s STARTDATE, --startDate STARTDATE
start date in iso format YYYY-MM-DD or 'firstDate'
which sets start date to first date in metadata
(default: 2020-03-01)
-e ENDDATE, --endDate ENDDATE
end date in iso format YYYY-MM-DD or 'lastDate' which
sets end date as last date in metadata (default:
lastDate)
Options passed to epimuller-draw:
-mt MINTIME, --MINTIME MINTIME
minimum time point to start plotting (default: 30)
-min MINTOTALCOUNT, --MINTOTALCOUNT MINTOTALCOUNT
minimum total count for group to be included (default:
50)
-c CASES_NAME, --cases_name CASES_NAME
file with cases - formated with 'date' in ISO format
and 'confirmed_rolling' cases, in tsv format (default:
None)
-l {date,time}, --xlabel {date,time}
Format of x axis label: ISO date format or timepoints
from start (default: date)
-lp {Right,Max,Start,End}, --labelPosition {Right,Max,Start,End}
choose position of clade labels (default: Right)
Options passed to epimuller-draw for page setup:
--WIDTH WIDTH WIDTH of page (px) (default: 1500)
--HEIGHT HEIGHT HEIGHT of page (px) (default: 1000)
--LEGENDWIDTH LEGENDWIDTH
LEGENDWIDTH to the right of plotting area (px)
(default: 220)
--MARGIN MARGIN MARGIN around all sides of plotting area (px)
(default: 60)
--FONTSIZE FONTSIZE
--LABELSHIFT LABELSHIFT
epimuller-define: make abundance and hiearchy files
epimuller-define [-h] (-n INNEXTSTRAIN | -a ANNOTATEDTREE) -m
INMETA [-p INPANGOLIN] [--noPangolin]
[-f TRAITOFINTERSTFILE] [-g GENEBOUNDRY]
[-k TRAITOFINTERSTKEY]
[-mut VOCLIST [VOCLIST ...]]
[-oDir OUTDIRECTORY] -oP OUTPREFIX
[-t TIMEWINDOW] [-s STARTDATE] [-e ENDDATE]
optional arguments:
-h, --help show this help message and exit
-n INNEXTSTRAIN, --inNextstrain INNEXTSTRAIN
nextstrain results with tree.nwk and
[traitOfInterst].json (default: None)
-a ANNOTATEDTREE, --annotatedTree ANNOTATEDTREE
nexus file name (default: None)
-m INMETA, --inMeta INMETA
metadata tsv with 'strain' and 'date'cols, optional:
cols of trait of interst; and pangolin col named:
'lineage' or 'pangolin_lin' (default: None)
-p INPANGOLIN, --inPangolin INPANGOLIN
pangolin output lineage_report.csv file, if argument
not supplied looks in inMeta for col with
'pangolin_lin' or 'lineage' (default: metadata)
--noPangolin do not add lineage to cade names (default: False)
-f TRAITOFINTERSTFILE, --traitOfInterstFile TRAITOFINTERSTFILE
name of nextstrain [traitOfInterst].json in
'inNextstrain' folder (default: aa_muts.json)
-g GENEBOUNDRY, --geneBoundry GENEBOUNDRY
json formated file specifing start end postions of
genes in alnment for annotatedTree with aa_muts option
(default: None)
-k TRAITOFINTERSTKEY, --traitOfInterstKey TRAITOFINTERSTKEY
key for trait of interst in json file or annotated
tree file for aa with 'mutations' annotation, use
'aa_muts', see example data/geneAAboundries.json
(default: aa_muts)
-mut VOCLIST [VOCLIST ...], --VOClist VOCLIST [VOCLIST ...]
list of aa of interest in form
[GENE][*ORAncAA][site][*ORtoAA] ex. S*501*, gaps
represed by X (default: None)
-oDir OUTDIRECTORY, --outDirectory OUTDIRECTORY
folder for output (default: ./)
-oP OUTPREFIX, --outPrefix OUTPREFIX
prefix of out files withen outDirectory (default:
None)
-t TIMEWINDOW, --timeWindow TIMEWINDOW
number of days for sampling window (default: 7)
-s STARTDATE, --startDate STARTDATE
start date in iso format YYYY-MM-DD or 'firstDate'
which is in metadata (default: 2020-03-01)
-e ENDDATE, --endDate ENDDATE
end date in iso format YYYY-MM-DD or 'lastDate' which
is in metadata (default: lastDate)
epimuller-draw: plot
epimuller-draw [-h] -p PARENTHIERARCHY_NAME -a ABUNDANCE_NAME
[-c CASES_NAME] -o OUTFOLDER [-mt MINTIME]
[-min MINTOTALCOUNT] [-l {date,time}]
[-lp {Right,Max,Start,End}] [--WIDTH WIDTH]
[--HEIGHT HEIGHT] [--LEGENDWIDTH LEGENDWIDTH]
[--LABELSHIFT LABELSHIFT] [--MARGIN MARGIN]
[--FONTSIZE FONTSIZE]
optional arguments:
-h, --help show this help message and exit
-p PARENTHIERARCHY_NAME, --parentHierarchy_name PARENTHIERARCHY_NAME
csv output from mutationLinages_report.py with child
parent col (default: None)
-a ABUNDANCE_NAME, --abundance_name ABUNDANCE_NAME
csv output from mutationLinages_report.py with
abundances of clades (default: None)
-c CASES_NAME, --cases_name CASES_NAME
file with cases - formated with 'date' in ISO format
and 'confirmed_rolling' cases, in tsv format (default:
None)
-o OUTFOLDER, --outFolder OUTFOLDER
csv output from mutationLinages_report.py with child
parent col (default: None)
-mt MINTIME, --MINTIME MINTIME
minimum time point to start plotting (default: 30)
-min MINTOTALCOUNT, --MINTOTALCOUNT MINTOTALCOUNT
minimum total count for group to be included (default:
50)
-l {date,time}, --xlabel {date,time}
Format of x axis label: ISO date format or timepoints
from start (default: date)
-lp {Right,Max,Start,End}, --labelPosition {Right,Max,Start,End}
choose position of clade labels (default: Right)
Options for page setup:
--WIDTH WIDTH WIDTH of page (px) (default: 1500)
--HEIGHT HEIGHT HEIGHT of page (px) (default: 1000)
--LEGENDWIDTH LEGENDWIDTH
LEGENDWIDTH to the right of plotting area (px)
(default: 220)
--LABELSHIFT LABELSHIFT
nudge label over by LABELSHIFT (px) (default: 15)
--MARGIN MARGIN MARGIN around all sides of plotting area (px)
(default: 60)
--FONTSIZE FONTSIZE
Install methods
With Bioconda
conda install -c bioconda epimuller
With pip
pip3 install epimuller
#If there is an issue with cairo, try:
pip3 install pycairo
pip3 install epimuller
From source
Download source code from gitHub or pypi
#open as needed for download format
tar -zxvf epimuller-[version].tar.gz
cd epimuller-[version]
python3 setup.py install
Run scripts directly
Note you will have to install all dependencies.
Download source code from gitHub or pypi
#open as needed for download format
tar -zxvf epimuller-[version].tar.gz
cd epimuller-[version]
#to run epimuller
python3 ./scripts/mutationLinages_report.py [arugments]
#to run epimuller-parse
python3 ./scripts/parseFastaNames.py [arugments]
#to run epimuller-define
python3 ./scripts/defineAndCountClades.py [arugments]
#to run epimuller-draw
python3 ./scripts/drawMuller.py [arugments]
Citation
Please link to this github if you have used epimuller in your research.
Extra notes on GISAID
If you do use GISAID data please acknowledge the contributers, such as with language suggested by GISAID.
