rafm Reliable AlphaFold Measures





rafm computes per-model measures such as expected global LDDT associated with atomic-level accuracy for AlphaFold models from pLDDT confidence scores.
Installation
You can install rafm via pip from PyPI:
$ pip install rafmUsage
rafm --help lists all commands. Current commands are:
-
- plddt-stats
-
Calculate stats on bounded pLDDTs from list of AlphaFold model files. in either PDB or mmCIF format.
Options:
-
- --criterion FLOAT
- The cutoff value on truncated pLDDT for possible utility. [default: 91.2]
-
- --min-length INTEGER
- The minimum sequence length for which to calculate truncated stats. [default: 20]
-
- --min-count INTEGER
- The minimum number of truncated pLDDT values for which to calculate stats [default: 20]
-
- --lower-bound INTEGER
- The pLDDT value below which stats will not be calculated. [default: 80]
-
- --upper-bound INTEGER
- The pLDDT value above which stats will not be calculated. [default: 100]
-
- --file-stem TEXT
- Output file name stem. [default: rafm]
Output columns (where NN is the bounds specifier, default: 80):
-
- residues_in_pLDDT
- The number of residues in the AlphaFold model.
-
- pLDDT_mean
- The mean value of pLDDT over all residues.
-
- pLDDT_median
- The median value of pLDDT over all residues.
-
- pLDDTNN_count
- The number of residues within bounds.
-
- pLDDTNN_frac
- The fraction of pLDDT values within bounds, if the count is greater than the minimum.
-
- pLDDTNNN_mean
- The mean of pLDDT values within bounds, if the count is greater than the minimum.
-
- pLDDTNN_median
- The median of pLDDT values within bounds, if the count is greater than the minimum.
-
- LDDT_expect
- The expectation value of global LDDT over the residues with LDDT within bounds. Only produced if default bounds are used.
-
- passing
- True if the model passed the criterion, False otherwise. Only produced if default bounds are used.
-
- file
- The path to the model file.
-
-
- plddt-select-residues
-
Writes a tab-separated file of residues from passing models, using an input file of values selected by plddt-stats. Input options are the same as plddt-stats.
Output columns:
-
- file
- Path to the model file.
-
- residue
- Residue number, starting from 0 and numbered sequentially. Note that all residues will be written, regardless of bounds set.
-
- pLDDT
- pLDDT value for that residue.
-
-
- plddt-plot-dists
-
Plot the distributions on the bounded pLDDT and residues in models that pass the selection criteria.
- Input Options:
-
-
- out-file-type
- Plot file extension of a type that matplotlib understands, (e.g., 'jpg', 'pdf') [default: png]
-
- residue-criterion
- Per-residue cutoff on usability (for plot only).
-
- Outputs:
-
When applied to set of "dark" genomes with no previous PDB entries, the distributions of median pLDDT scores with a lower bound of 80 and per-residue pLDDT scores with a minimum of 80 looks like this:
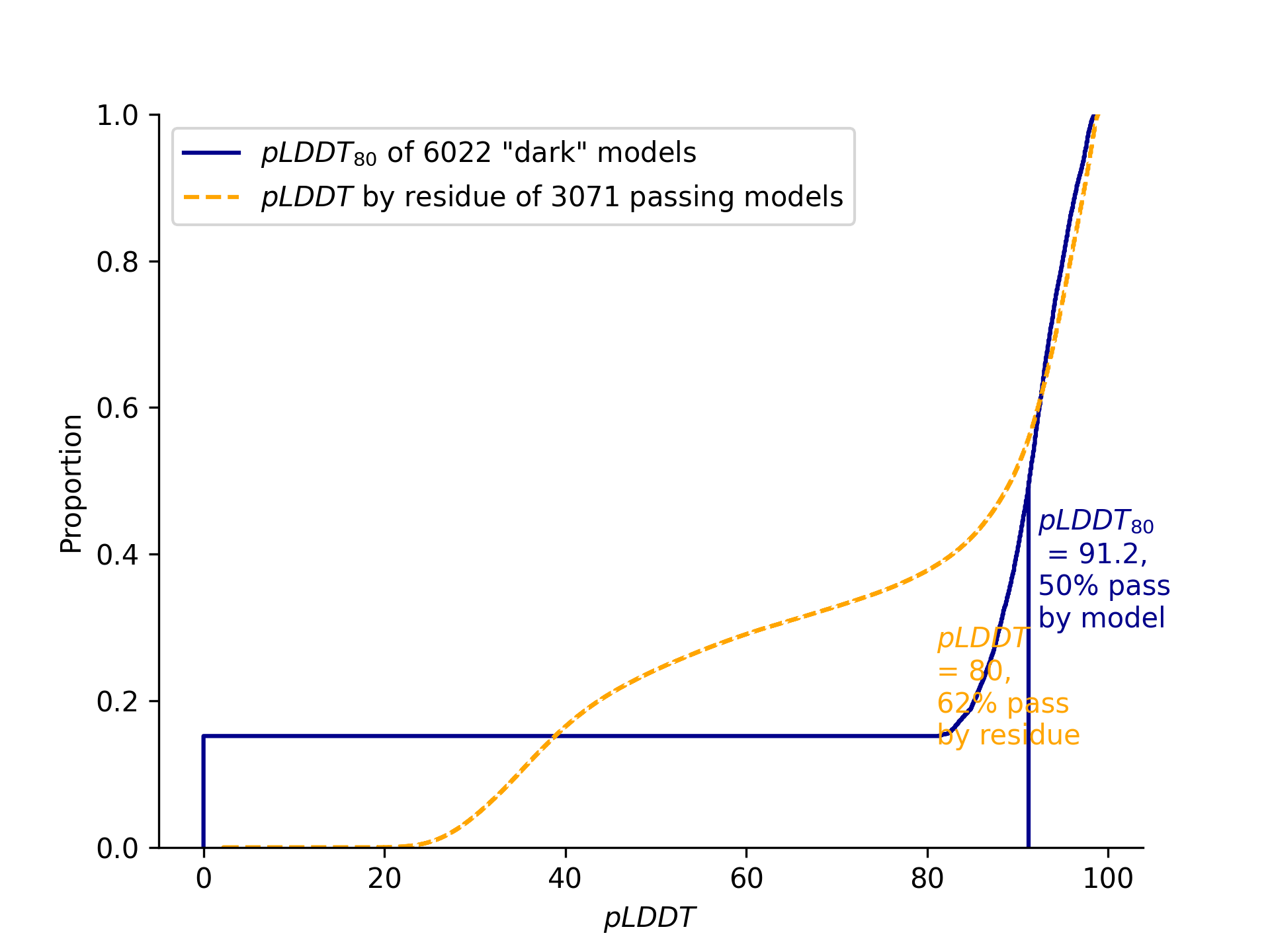
-
- stats
-
Produce a set of summary stats on results of runs. See also the global stats file rafm_stats.json.
Statistical Basis
The default parameters were chosen to select for LDDT values of greater than 80 on a set of crystal structures obtained since AlphaFold was trained. The distributions of LDDT scores for the passing and non-passing sets, along with an (overlapping) set of AlphaFold model files at 100% sequence identity over at least 80% of the sequence looks like this:
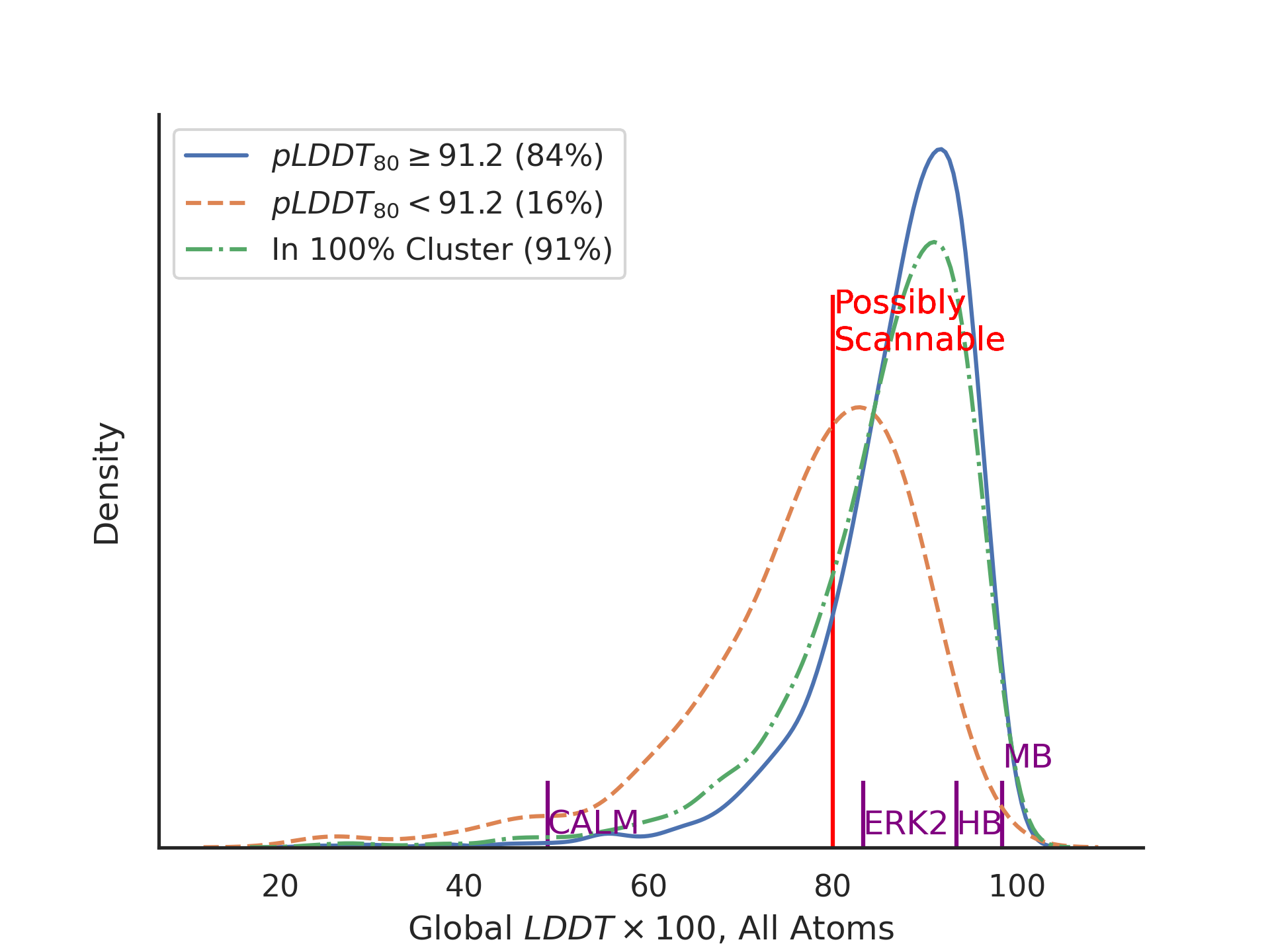
The markers on the x-axis refer to the size of conformational changes observed in conformational changes in various protein crystal structures:
-
- CALM
- Between calcium-bound and calcium-free calmodulin (depicted in the logo image above).
-
- ERK2
- Between unphosphorylated and doubly-phosphorylated ERK2 kinase.
-
- HB
- Between R- and T-state hemoglobin
-
- MB
- Between carbonmonoxy- and deoxy-myoglobin
The value of LDDT >= 80 we selected as the minimum value that was likely to prove useful for virtual screening. The per-residue value of pLDDT >= 80 was also chosen as the minimum likely to give the correct side-chain rotamers for a surface defined by contacts between two residues. A choice of 91.2 as a criterion leads to the following confusion matrix versus a set of post-training crystal structures:
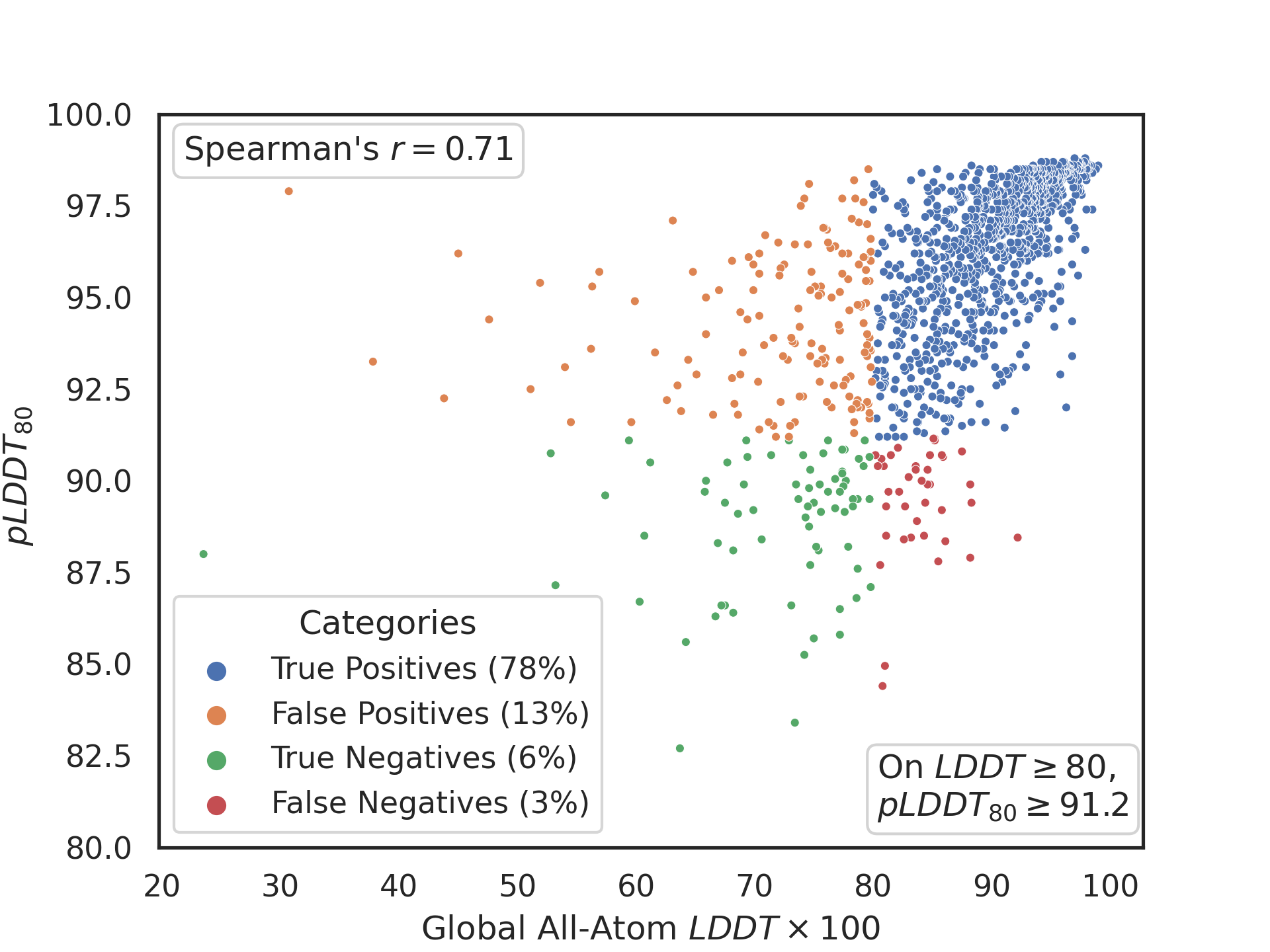
At a correlation coefficient of 0.71, this correlation isn't great, but enough to demonstrate a usable sensitivity. After we fix a few problems with the alignments, it may go a bit higher but our feeling is probably not more than about 0.8. The support will get better, but the criterion on this metric seems unlikely to change.
Contributing
Contributions are very welcome. To learn more, see the Contributor Guide.
License
Distributed under the terms of the MIT license, rafm is free and open source software.
Issues
If you encounter any problems, please file an issue along with a detailed description.
Credits
This project was generated from the UNM Translational Informatics Python Cookiecutter template.
rafm was written by Joel Berendzen and Jessica Binder.